
Minden iLive-tartalmat orvosi szempontból felülvizsgáltak vagy tényszerűen ellenőriznek, hogy a lehető legtöbb tényszerű pontosságot biztosítsák.
Szigorú beszerzési iránymutatásunk van, és csak a jó hírű média oldalakhoz, az akadémiai kutatóintézetekhez és, ha lehetséges, orvosilag felülvizsgált tanulmányokhoz kapcsolódik. Ne feledje, hogy a zárójelben ([1], [2] stb.) Szereplő számok ezekre a tanulmányokra kattintható linkek.
Ha úgy érzi, hogy a tartalom bármely pontatlan, elavult vagy más módon megkérdőjelezhető, jelölje ki, és nyomja meg a Ctrl + Enter billentyűt.
Charcot-Marie-Tooth betegség.
A cikk orvosi szakértője

Utolsó ellenőrzés: 04.07.2025
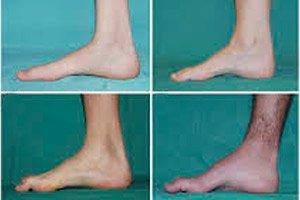
A peroneális izomsorvadás, Charcot-Marie-Tooth szindróma vagy betegség krónikus örökletes betegségek egy csoportja, amelyek a perifériás idegek károsodásával járnak.
Az ICD-10 szerint az idegrendszeri betegségekről szóló részben a betegség kódja G60.0 (örökletes motoros és szenzoros neuropátia). Az árva betegségek listáján is szerepel.
Járványtan
A klinikai statisztikák szerint a Charcot-Marie-Tooth betegség minden típusának prevalenciája 100 ezer lakosra vetítve 19 eset (más források szerint egy eset 2,5-10 ezer lakosra vetítve).
Az 1-es típusú CMT az esetek körülbelül kétharmadát teszi ki (5000-7000 lakosra jut egy eset), és ezek közel 70%-a a PMP22 gén duplikációjával jár. Világszerte több mint 1,2 millió ember szenved ebben a típusú betegségben.
A 4-es típusú CMT előfordulása becslések szerint 10 000 gyermekre vetítve 1-5 eset. [ 1 ]
Okoz Charcot-Marie-Tooth betegség
A polineuropátiás szindrómák osztályozása szerint a peroneális (fibuláris) izomatrófia, a Charcot-Marie-Tooth neurális amiotrófia vagy Charcot-Marie-Tooth betegség (rövidítve CMT) genetikailag meghatározott motoros-szenzoros polineuropátiákra utal. [ 2 ]
Vagyis előfordulásának okai genetikai mutációk. A genetikai eltérések jellegétől függően a szindróma fő típusait vagy fajtáit különböztetjük meg: demyelinizációs és axonális. Az első csoportba tartozik az 1-es típusú Charcot-Marie-Tooth betegség (CMT1), amely a PMP22 gén 17-es kromoszómán történő duplikációja miatt következik be, amely a transzmembrán perifériás mielinfehérjét 22 kódolja. Ennek eredményeként az axonhüvely (idegsejt-nyúlványok) szegmentális demyelinizációja és az idegjel vezetési sebességének csökkenése következik be. Ezenkívül mutációk lehetnek más génekben is.
Az axonális forma a 2-es típusú Charcot-Marie-Tooth betegség (CMT2), amely magukat az axonokat érinti, és az MFN2 gén 1p36.22 lókuszában található kóros elváltozásaival jár, amely a mitofusin-2 membránfehérjét kódolja, amely szükséges a mitokondriumok fúziójához és a perifériás idegsejtekben található funkcionális mitokondriális hálózatok kialakulásához. A CMT2-nek több mint egy tucat altípusa létezik (specifikus génmutációkkal).
Megjegyzendő, hogy jelenleg több mint száz olyan gént azonosítottak, amelyek öröklődő károsodása a Charcot-Marie-Tooth betegség különböző altípusait okozza. Például az RAB7 gén mutációi a 2B típusú CMT-hez vezetnek; az SH3TC2 gén (amely a Schwann-sejtek egyik membránfehérjéjét kódolja) megváltozása a 4C típusú CMT-t okozza, amely gyermekkorban jelentkezik, és a motoros és szenzoros neuronok demielinizációja jellemzi (a betegség 4-es típusának másfél tucat formája létezik).
Egy ritka 3-as típusú CMT (Dejerine-Sottas szindróma) kora gyermekkorban kezd kialakulni, és a PMP22, MPZ, EGR2 és más gének mutációi okozzák.
Amikor az 5-ös típusú CMT 5-12 éves korban jelentkezik, nemcsak a motoros neuropátia (az alsó végtagok spasztikus paraparézise formájában) figyelhető meg, hanem a látóideg és a hallóidegek károsodása is.
Az izomgyengeség és a látóideg-sorvadás (látásvesztéssel), valamint az egyensúlyproblémák jellemzőek a 6-os típusú CMT-re. A 7-es típusú Charcot-Marie-Tooth betegségben nemcsak motoros-szenzoros neuropátia, hanem retinitis pigmentosa formájában jelentkező retinabetegség is jelentkezik.
A férfiaknál gyakoribb az X-kromoszómához kötött CMT vagy Charcot-Marie-Tooth betegség, amely a végtagok tetraparézisével (mindkét kar és láb mozgásgyengesége) jár, egy demyelinizációs típus, és feltehetően az X kromoszóma hosszú karján található GJB1 gén mutációjából ered, amely a connexin 32-t kódolja, a Schwann-sejtek és az oligodendrociták transzmembrán fehérjéjét, amely az idegjelek továbbítását szabályozza. [ 3 ]
Kockázati tényezők
A CMT fő kockázati tényezője a betegség családi előfordulása, azaz a közeli hozzátartozóknál való előfordulása.
A genetikusok szerint, ha mindkét szülő hordozza a Charcot-Marie-Tooth betegség autoszomális recesszív génjét, akkor a betegség kialakulásának kockázata 25%. Annak a kockázata, hogy a gyermek hordozza ezt a gént (de nem lesznek tünetei), 50%-ra becsülhető.
X-kromoszómához kötött öröklődés esetén (amikor a mutált gén a nő X kromoszómáján található) 50% a kockázata annak, hogy az anya átadja a gént a fiának, akinél CMT alakul ki. Előfordulhat, hogy a betegség nem lánygyermek születésekor jelentkezik, de a lány fiai (unokái) örökölhetik a hibás gént, és a betegség kialakulhat.
Pathogenezis
Bármely Charcot-Marie-Tooth betegség esetén a patogenezisét a perifériás idegek örökletes rendellenessége okozza: motoros (mozgási) és szenzoros (érzékeny).
Ha a CMT típus demyelinizáló, akkor a perifériás idegek axonjait védő mielinhüvely pusztulása vagy hibája az idegimpulzusok átvitelének lassulásához vezet a perifériás idegrendszerben - az agy, az izmok és az érzékszervek között.
A betegség axonális típusában maguk az axonok érintettek, ami negatívan befolyásolja az idegjelek erősségét, ami nem elegendő az izmok és az érzékszervek teljes stimulálásához.
Olvasd el ezt is:
Hogyan terjed a Charcot-Marie-Tooth szindróma? A hibás gének autoszomális domináns vagy autoszomális recesszív módon öröklődhetnek.
A leggyakoribb típus, az autoszomális domináns öröklődésmód akkor fordul elő, amikor a mutált génből csak egy példány létezik (az egyik szülő hordozza). A CMT minden született gyermekre való átadásának valószínűsége 50%-ra becsülhető [ 4 ].
Autoszomális recesszív öröklődésmód esetén a hibás gén két példánya (mindkét szülőtől egy, akik nem mutatják a betegség tüneteit) szükséges a betegség kialakulásához.
Az esetek 40-50%-ában autoszomális domináns módon öröklődő demyelinizáció, azaz 1-es típusú CMT fordul elő; az esetek 12-26%-ában axonális CMT, azaz 2-es típusú. Az esetek 10-15%-ában pedig X-kromoszómához kötött öröklődés figyelhető meg. [ 5 ]
Tünetek Charcot-Marie-Tooth betegség
A betegség első jelei jellemzően gyermekkorban és serdülőkorban kezdenek megjelenni, és fokozatosan fejlődnek az egész életen át, bár a szindróma később is jelentkezhet. A tünetek kombinációja változó, és a betegség progressziójának üteme, valamint súlyossága lehetetlen megjósolni.
A kezdeti stádium tipikus tünetei közé tartozik a fokozott általános fáradtság; a láb, a boka és a sípcsont izmainak csökkent tónusa (gyengesége); a reflexek hiánya. Ez megnehezíti a lábmozgást, és diszbáziához (járási zavarhoz) vezet, amely a lábak magasabbra emelkedésében jelentkezik, gyakran gyakori botlásokkal és esésekkel. A Charcot-Marie-Tooth betegség jelei kisgyermekeknél a kifejezett ügyetlenség és a kortól eltérő járási nehézségek lehetnek, amelyek kétoldali lábleeséssel járnak. A lábdeformitások is jellemzőek: magas lábboltozat (beesett láb) vagy súlyos lúdtalp, görbe (kalapács alakú) lábujjak.
Izomhypotonia hátterében lábujjhegyen járás esetén a neurológus gyaníthatja, hogy a gyermeknek 4-es típusú CMT-je van, amelyben a gyerekek serdülőkorukra nem tudnak járni.
A betegség előrehaladtával az izomsorvadás és -gyengeség a felső végtagokra is átterjed, ami megnehezíti a finommotoros készségek ellátását és a szokásos kézműveletek elvégzését. A tapintási érzékelés és a hő- és hidegérzékelés képességének csökkenése, valamint a lábak és a kezek zsibbadása az érzőidegek axonjainak károsodására utal.
A gyermekkorban jelentkező 3-as és 6-os típusú Charcot-Marie-Tooth betegség esetén szenzoros ataxia (a mozgások és az egyensúly koordinációjának zavara), izomrángás és remegés, az arcideg károsodása, látóideg-sorvadás nystagmussal, valamint halláskárosodás figyelhető meg.
Későbbi stádiumokban kontrollálhatatlan remegés (tremor) és gyakori izomgörcsök jelentkezhetnek; a mozgási problémák izom-, ízületi és neuropátiás fájdalmak kialakulásához vezethetnek.
Komplikációk és következmények
A Charcot-Marie-Tooth betegségnek lehetnek szövődményei és következményei, mint például:
- gyakoribb rándulások és törések;
- a periartikuláris izmok és inak rövidülésével járó kontraktúrák;
- szkoliózis (a gerinc görbülete);
- légzési problémák – amikor a rekeszizom izmait beidegző idegrostok károsodnak:
- az önálló mozgás képességének elvesztése.
Diagnostics Charcot-Marie-Tooth betegség
A diagnózis magában foglalja a klinikai vizsgálatot, az anamnézist (beleértve a családi kórtörténetet), a neurológiai és szisztémás vizsgálatot.
A vizsgálatokat a mozgástartomány, az érzékenység és az ínreflexek ellenőrzésére végzik. Az idegvezetés mértékét műszeres diagnosztikai módszerekkel – elektromiográfiával vagy elektroneuromiográfiával – lehet értékelni. Ultrahangra vagy MRI-re is szükség lehet. [ 6 ]
A CMT-t okozó leggyakoribb genetikai mutációk vérmintában történő kimutatására szolgáló genetikai vagy DNS-vizsgálat korlátozott, mivel a DNS-tesztek jelenleg nem állnak rendelkezésre a betegség minden típusára. További információkért lásd: Genetikai vizsgálatok
Bizonyos esetekben a perifériás ideg (általában a suralis ideg) biopsziáját végzik.
Megkülönböztető diagnózis
A differenciáldiagnózis magában foglalja az egyéb perifériás neuropátiákat, a Duchenne-izomdisztrófiát, a mielopátiás és myastheniás szindrómákat, a diabéteszes neuropátiát, a multiplex és amiotrófiás laterális szklerózis myelopathiáit, a Guillain-Barré-szindrómát, a szárideg traumáját és sorvadását (beleértve a gerinc ágyéki porckorongjai közé történő becsípődést is), a kisagy vagy a talamusz károsodását, valamint a kemoterápia mellékhatásait (citosztatikumokkal, például vinkrisztinnel vagy paklitaxellel végzett kezelés során). [ 7 ]
Ki kapcsolódni?
Kezelés Charcot-Marie-Tooth betegség
Ennek az örökletes betegségnek a kezelése ma már magában foglalja a gyógytornát (amelynek célja az izmok erősítése és nyújtása); a foglalkozásterápiát (amely segíti a kézizomgyengeségben szenvedő betegeket); és ortopédiai eszközök használatát a járás megkönnyítése érdekében. Szükség esetén fájdalomcsillapítókat vagy görcsoldókat írnak fel. [ 8 ]
Súlyos lúdtalp esetén oszteotómiát lehet végezni, sarokdeformáció esetén pedig sebészeti korrekciót – artrodézist – javasolnak. [ 9 ]
Jelenleg is folynak kutatások a betegség genetikai komponensével és kezelési módszereivel kapcsolatban. Az őssejtek, bizonyos hormonok, lecitin vagy aszkorbinsav alkalmazása eddig nem hozott pozitív eredményeket.
A legújabb kutatásoknak köszönhetően azonban a közeljövőben valóban megjelenhet valami új a Charcot-Marie-Tooth betegség kezelésében. Így 2014 óta a francia Pharnext cég fejleszti, 2019 közepe óta pedig klinikai vizsgálatok folynak a felnőttek 1-es típusú CMT-jének kezelésére szolgáló PXT3003 gyógyszerrel, amely gátolja a PMP22 gén fokozott expresszióját, javítja a perifériás idegek mielinizációját és enyhíti a neuromuszkuláris tüneteket.
A Sarepta Therapeutics (USA) orvosi vállalat szakemberei egy génterápiás kezelésen dolgoznak az 1-es típusú Charcot-Marie-Tooth betegség kezelésére. Ez a terápia egy ártalmatlan, lineáris, egyszálú DNS-genomú Dependovirus nemzetségbe tartozó adeno-asszociált vírust (AAV) használ, amely az NTF3 gént juttatja be a szervezetbe, amely a Schwann-idegsejtek működéséhez szükséges neurotrophin-3 (NT-3) fehérjét kódolja.
A Helixmith 2020 végéig megkezdi a dél-koreai fejlesztésű Engensis (VM202) génterápia klinikai vizsgálatait az 1-es típusú CMT izomtüneteinek kezelésére. [ 10 ]
Megelőzés
A CMT megelőzése a leendő szülők genetikai tanácsadásával történhet, különösen, ha a párban valakinek családi előfordulása van a betegséggel. Azonban de novo pontgénmutációk eseteit is azonosították, azaz a betegség hiányában a családi kórtörténetben.
Terhesség alatt a Charcot-Marie-Tooth betegség valószínűségét a jövőbeli gyermekben chorionboholy biopsziával (a terhesség 10. és 13. hete között), valamint a magzatvíz elemzésével (15-18 héten) lehet ellenőrizni.
Előrejelzés
Általánosságban elmondható, hogy a Charcot-Marie-Tooth betegség különböző típusainak prognózisa a klinikai súlyosságtól függ, de minden esetben a betegség lassan halad. Sok betegnél fogyatékosság tapasztalható, bár ez nem csökkenti a várható élettartamot.